
Kalıtsal hastalık
- Kalıtım örnekleri

Otozomal dominant kalıtım
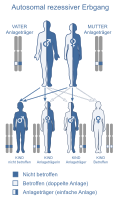
Otozomal resesif kalıtım


Bir şekilde kalıtsal hastalık (ya da genetik hastalık ) hastalıkları ve özel özellikleri yoluyla tayin edilir mutasyon bir de (gen varyantı) geni ( tek gen ) ya da (farklı genlerde fazla mutasyon (gen varyantları) poligenik tetiklenebilir) ve bazı Erkrankungs de eğilimleri önderlik etmek. Bu bağlamda, monojenik veya poligenetik hastalıktan söz edilir .
Ancak daha dar anlamda, başlangıçtan itibaren atipik olarak değişen genlerin tetiklediği ve kalıtım yoluyla atalarından yavrularına aktarılan kalıtsal hastalıklar arasında sadece bu hastalıklar ve özellikler sayılır . Kalıtım yollarını araştırmanın en eski yöntemi , örneğin hemofili veya renk körlüğünün daha sık meydana geldiği aile ağaçlarının soyağacı analiziydi .
Genelde 46 kromozomun insan genomunda bulunmadığı trizomi formları gibi sendromlar , kesin olarak söylemek gerekirse, kalıtsal bir hastalık olarak sayılamaz, çünkü bunlar genellikle yalnızca embriyonun hücreleri bölündüğünde kendiliğinden ortaya çıkar. bu nedenle nadiren bir ebeveynden miras alınır.
Farklı şekiller
Kalıtsal hastalıklar farklı kalıtım modellerini takip eder ve farklı kalıtım, nüks ve hastalık olasılıkları ile ilişkilidir. Otozomal resesif ve gonozomal ve mitokondriyal kalıtımdan otozomal dominant kalıtım arasında bir ayrım yapılır .
Otozomal resesif kalıtım

Tuhaflık yalnızca, her iki kromozomdaki belirli bir genin her iki kopyasında da bir değişiklik ( mutasyon ) varsa ortaya çıkar , yani. Yani, söz konusu kişi biyolojik babasından ve biyolojik annesinden bir değişiklik miras almışsa. Ebeveynlerin etkilenmesi gerekmez, bu nedenle fenotip her nesilde ortaya çıkmaz. Mutasyonun aynı olması gerekmez. Moleküler genetik ile ayırt edilebilen iki mutasyon, bir gende aynı işlev kaybına yol açıyorsa, bileşik heterozigotluktan söz edilir . Otozomal resesif kalıtım örnekleri kistik fibroz , albinizm ve fenilketonüridir (PKU) (fenilalanin hidroksilazda bir kusur).
Otozomal resesif kalıtımsal hastalıklar çoğunlukla işlev kaybı mutasyonlarıdır . Otozomal resesif kalıtımdan belirgin sapmaların nedenleri, sözde baskınlık , heterojenlik , izodizomi ve hasta çocuklu heterozigotların hesaplamaya dahil edilmediği gerçeğidir . Tipik örnekler:
- Adrenogenital sendrom (AGS),
- Akçaağaç şurubu hastalığı ,
- Albinizm ,
- Alkaptonüri ,
- Alfa1-antitripsin eksikliği ,
- Galaktozemi ,
- Kalıtsal fruktoz intoleransı
- Hemokromatoz
- Joubert sendromu ,
- Kretinizm ,
- Kısa kaburga polidaktili sendromları (tip I, II, III, IV),
- Laurence-Moon-Biedl-Bardet sendromu ( LMBB sendromu ),
- Yarık dudak ve damak
- Wilson hastalığı
- Mukopolisakkaridozlar (MPS),
- Kistik fibroz veya kistik fibroz ,
- Fin tipi nefrotik sendrom ,
- Peters Plus Sendromu ,
- Fenilketonüri (PKU),
- Ribbing sendromu ,
- Talasemi ve
- Xeroderma pigmentosum .
- Otozomal resesif polikistik böbrek hastalığı (ARPKD)
Otozomal dominant kalıtım

Burada iki homolog kromozomdan birinde değiştirilmiş bir alel (aleller, bir diploid kromozom kümesinin karşılıklı ve aynı zamanda karşıt olarak karşılık gelen genleridir ) karakteristik ifadeye yol açar. Genetik bilgi 44 otozomdan birinde mevcuttur ve cinsiyete bakılmaksızın miras alınır. Yani kadınlar ve erkekler eşit derecede etkileniyor. Fenotip her nesil meydana gelir. Örnekler:
- Akondroplazi
- Apert sendromu
- Brakidaktili
- Huntington Hastalığı ("Aziz Vitus Dansı")
- Ehlers-Danlos sendromu (tip I - IV, VII A / B, VIII)
- Engelmann sendromu
- Eritropoietik protoporfiri
- Faktör V Leiden mutasyonu
- Ailevi hiperkolesterolemi
- HMSN tip I ( Charcot-Marie-Tooth hastalığı )
- Kötü huylu hipertermi
- Marfan Sendromu
- Darier hastalığı
- Çoklu kıkırdaklı ekzostozlar
- Tip I miyotonik distrofi
- Nörofibromatozis (Recklinghausen hastalığı)
- Osteogenezis imperfekta (tip I)
- Piebaldizm
- Polidaktili
- Retinoblastom
- Ruvalcaba-Myhre-Smith sendromu ve
- Orak hücre anemisi
- Tüberoskleroz
- Otozomal Dominant Polikistik Böbrek Hastalığı (ODPBH)
Gonozomal kalıtım
Kalıtsal gonozomal hastalıklar, yani değişikliğin cinsiyet kromozomları X veya Y'yi etkilediği durumlar , Y kromozomu daha az gen içerdiğinden çoğu durumda X kromozomu üzerindedir . X kromozomu 155 megabaza, Y kromozomu 59 megabaza sahiptir. X'e bağlı kalıtım örneğini kullanarak, aşağıdaki özellikler netleşir:
X'e bağlı resesif
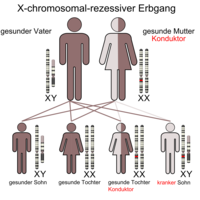
X'e bağlı resesif kalıtım (anne bir taşıyıcıdır)
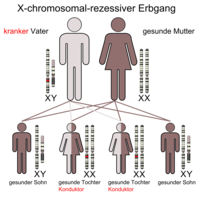
X'e bağlı resesif kalıtım (eğer baba hastaysa)


Kızlar / kadınlar yalnızca her iki X kromozomu da hasar gördüğünde etkilenir, aksi takdirde sadece taşıyıcıdırlar ( iletkenler ), d. Başka bir deyişle, değişen X kromozomunu çocuklarına aktarabilirler, ancak kendileri buna karşılık gelen bir fenotip geliştirmezler. Kızlar / kadınlar, bir X kromozomundaki değişikliği, eğer değiştirilmezse, ikinci X kromozomu ile telafi edebilirler. Erkekler / erkekler, değiştirilmiş X kromozomlarından birini fenotipik olarak sağlıklı anneden miras alırlarsa veya fenotipik olarak hasta bir anneden iki değişmiş X kromozomundan birini alırlarsa etkilenirler, çünkü erkeklerin / erkeklerin kesinlikle bir anneden X kromozomu vardır ve sadece bu. Erkekler / erkekler fenotipik olarak daha sık etkilenir, çünkü kızlar / kadınlar kusuru diğer X kromozomu ile telafi eder. Örnekler, glikoz-6-fosfat dehidrojenaz eksikliği (G-6-PD eksikliği), hemofili A ve B (hemofili), Lesch-Nyhan sendromu , Fabry hastalığı , mukopolisakkaridoz tip II, kas distrofisi (tip Duchenne, tip Becker-Kiener), Norrie sendromu , retinitis pigmentosa , kırmızı-yeşil körlük , septik granülomatoz , X-SCID (şiddetli kombine bağışıklık eksikliği) ve ornitin transkarbamilaz (OTC) eksikliği ( üre döngüsü bozukluğu )
X'e bağlı baskın

X'e bağlı baskın miras (hasta babayla)

X'e bağlı baskın kalıtım (hasta annede)


Anneleri bir X kromozomunda hastalığa neden olan bir allel taşıyıcısı ise, erkek çocuklar / erkekler% 50 etkilenir . Öte yandan, X kromozomlarının her ikisi de hastalığa neden olan aleli içeriyorsa, tüm çocuklar etkilenir. Genel olarak, kızlar / kadınlar daha sık etkilenir çünkü değiştirilmiş bir X kromozomu alma olasılığı, iki X kromozomunda (biri babadan, biri anneden) erkeklerden / erkeklerden (biri anneden) daha yüksektir. Örnekler, ailesel fosfatemik raşitizm (aynı zamanda idiyopatik Debré-de-Toni-Fanconi sendromu veya D vitaminine dirençli raşitizm olarak da adlandırılır ), Rett sendromu ve orofacio-dijital sendrom tip 1'dir .
Mitokondriyal veya ekstrakromozomal kalıtım
Bir insan hücresindeki DNA'nın yaklaşık yüzde 0,1'i çekirdekte değil mitokondriyadadır . Yumurta hücreleri, spermin aksine birkaç yüz bin mitokondriye sahip olduğundan, mitokondriyal DNA'daki mutasyonlar sadece anne tarafında kalıtılır. Aynısı fotosentetik olarak aktif organizmaların kloroplastları için de geçerlidir .
Ekstrakromozomal kalıtıma da bakınız.
Teşhis ve tedavi
| ICD-10'a göre sınıflandırma | |
|---|---|
| Q90 - Q99 | Başka yerde sınıflandırılmamış kromozomal anormallikler |
| ICD-10 çevrimiçi (WHO versiyonu 2019) | |
Kalıtsal bir hastalıktan şüpheleniliyorsa, insan genetik testi netlik sağlayabilir. Kromozomlar, sayısal ve yapısal değişiklikler için kontrol edilir. Belirli bir genetik kusurun acil bir şüphesi varsa, tek tek gen takımyıldızlarının daha kapsamlı, karmaşık bir araştırması da mümkündür. Sonuçlar daha sonra kalıtım riskinin değerlendirilmesinde yardımcı olabilir.
Eğer genetik makyaj kendine mahsustur, bugünün tıbbi seçenekleri ile nedenlere hareket etme genellikle mümkün değildir. Bu nedenle, genellikle yaşam tarzı, risk faktörleri hakkında eğitim ve semptomatik önlemlerle ilgili tavsiyeler verilir . O zaman bunlar bireysel kararlardır, özellikle de her zaman bir hastalık değil, çoğu zaman bir eğilim olduğu için .
Gibi birkaç hastalık için B. spinal musküler atrofi, tedavide ilk girişimler var.
Tarih
Kalıtsal hastalık kavramı, sadece anlamında 20. yüzyıldan beri kullanılmaktadır genetik hastalık , oldu genellikle yanlış kullanılan ilk yarısında 20. yüzyılın böyle “suç eğilimlerinin” ya da “anti-toplumsallık olarak iddia edilen“hastalıklar”da dahil olmak üzere, ”. Bu düşünce kısırlaştırma programlarını ve ötenazi fikrini etkiledi ve aşırı ifadesini Alman Ulusal Sosyalizminde buldu , ancak o zamanlar ABD, İngiltere ve Fransa gibi diğer birçok ülkede de mevcuttu. Günümüzde sadece bu hastalıklara, olabildiğince açık bir şekilde tanımlanabilen ve büyük olasılıkla genetik kusurlara bağlı olan kalıtsal hastalıklar denmektedir.
Diğer kalıtsal hastalıklar ve özellikler
- Mayer-Rokitansky-Küster-Hauser sendromu
- Kalıtsal Spastik Spinal Felç (HSP / FSP)
- Hipofosfatazi
- İhtiyoz
- Kedi Gözü Sendromu
- Retinitis pigmentosa , Usher sendromu
- Tüberoskleroz
- Wolf-Hirschhorn Sendromu
Genetik olarak belirlenmiş eğilim
Klasik kalıtsal hastalık anlamında çeşitli hastalıklar, sakatlıklar ve özellikler kalıtsal değildir, ancak bunların ortaya çıkmasına (bazen ailesel) bir genetik hastalık yatkınlığı (yatkınlık, yatkınlık) neden olabilir. Bunlar şunları içerir: B.:
- Obezite
- Alerjiler , çeşitli
- Alzheimer hastalığı
- Otoimmün hastalıklar
- Bipolar bozukluk
- yüksek tansiyon
- Creutzfeldt-Jakob hastalığı
- depresyon
- Şeker hastalığı
- Ayak başparmağı deviasyonu ( Halluks valgus )
- Saç kaybı
- Kalp hastalığı
- Kalp krizi
- Çeşitli kanserler ( Alman Tabipler Birliği'nin web sitesinde kansere genetik yatkınlığın teşhisi için kılavuza bakın )
- Laktoz intoleransı
- kötü huylu hipertermi
- migren
- Çoklu skleroz (MS)
- kemik erimesi
- Parkinson hastalığı
- Sedef hastalığı
- romatizma
- şizofreni
- inme
- sağırlık
- Şekilleri Trizomi ( eğilim bir "varsa yavruda bir translokasyon trizami geliştirmek için dengeli translokasyon ebeveynlerde gelen kromozom" Trizomi ilgili formu olmadan)
- Vitiligo
Ayrıca bakınız
- Kalıtsal hastalıkların listesi
- genetik
- Kalıtsal kanserler
- Doğum öncesi tanı
- Preimplantasyon Tanıları
- Köpeklerin kalıtsal hastalıkları (kategori)
İnternet linkleri
Bireysel kanıt
- ↑ Ulrich Weber: Biyoloji üst seviyesi. Toplam bant. Cornelsen, Berlin 2001, ISBN 3-464-04279-0 , s. 180-182.
- ↑ Ensembl veritabanı , 11 Şubat 2017'de erişildi
- ↑ JE Wraith: Ornitin Karbamoiltransferaz eksikliği. In: Çocuklukta Hastalık Arşivleri . Ocak 2001, Cilt 84, Sayı 1, sayfa 84-88: Gözden Geçirme. PMID 11124797 .
- ↑ Werner oğlu: Kalıtsal hastalıklar. İçinde: Werner E. Gerabek , Bernhard D. Haage, Gundolf Keil , Wolfgang Wegner (editörler): Enzyklopädie Medizingeschichte. De Gruyter, Berlin / New York 2005, ISBN 3-11-015714-4 , sayfa 366 f .; burada: s. 366.
- ↑ Wolfgang Ayaß : "Asosyal çocuklar, ulusal topluluk için tamamen istenmeyen bir durumdur". Sosyal dışlayıcıların zorla kısırlaştırılması . İçinde: Margret Hamm (Ed.): Hayata değmez - hayatları mahvetti. Zorla kısırlaştırma ve "ötenazi". Verlag für Akademische Schriften (VAS), Frankfurt am Main 2005, ISBN 3-88864-391-0 , s. 111–119.